
-

����ͨ��
����ץס�����Ƽ�
���ӵ��}��
����ͨ��
����ץס�����Ƽ�
���ӵ��}��
�L�x�L�����M�M�b�e�`�\���c�������ԡ����Ŀ����������u��
��Nature Biotechnology����Troubleshooting common errors in assemblies of long-read metagenomes
�����w�� �� �� С �� �r�g��2026��01��03�� ��Դ��Nature Biotechnology 41.7
�����]��
�������о�ᘌ��L�x�L�����M�M�b�ʴ_���u���y�}���_�l�˻����x�μ����¼��������_Դ��������ϵ�y�u����HiCanu��hifiasm-meta��metaFlye��metaMDBG�Ŀ����21��PacBio HiFi�������ϵı��F���о��l�F�M�b�e�`�ʸ��_ÿ1�|�A����46���e�`����ʾ����Ƕ���w���^��h�����P�I���}����������y���g���������M�ؽ��еĿɿ����ṩ��Ҫ���ϡ�
�S���ڶ����y���g�İlչ���������M����ֱ�ӏĭh�������M���ؽ����o�����B���ɽ�ʾ����������c���ܡ�Ȼ�����x�L�y��ľ����Ԍ��»���M�M�b�߶���Ƭ���Ҵ�����Ⱦ���������y���g����PacBio��Oxford Nanopore��ͨ�^���L�x�Lͻ���؏��������ƣ�����s����Ⱥ���о������C�������L�x�L�M�b�㷨�ڏ��s�h���еđ��Üʴ_�������R�������䌦��ȱ����������M�ĵ��S�����
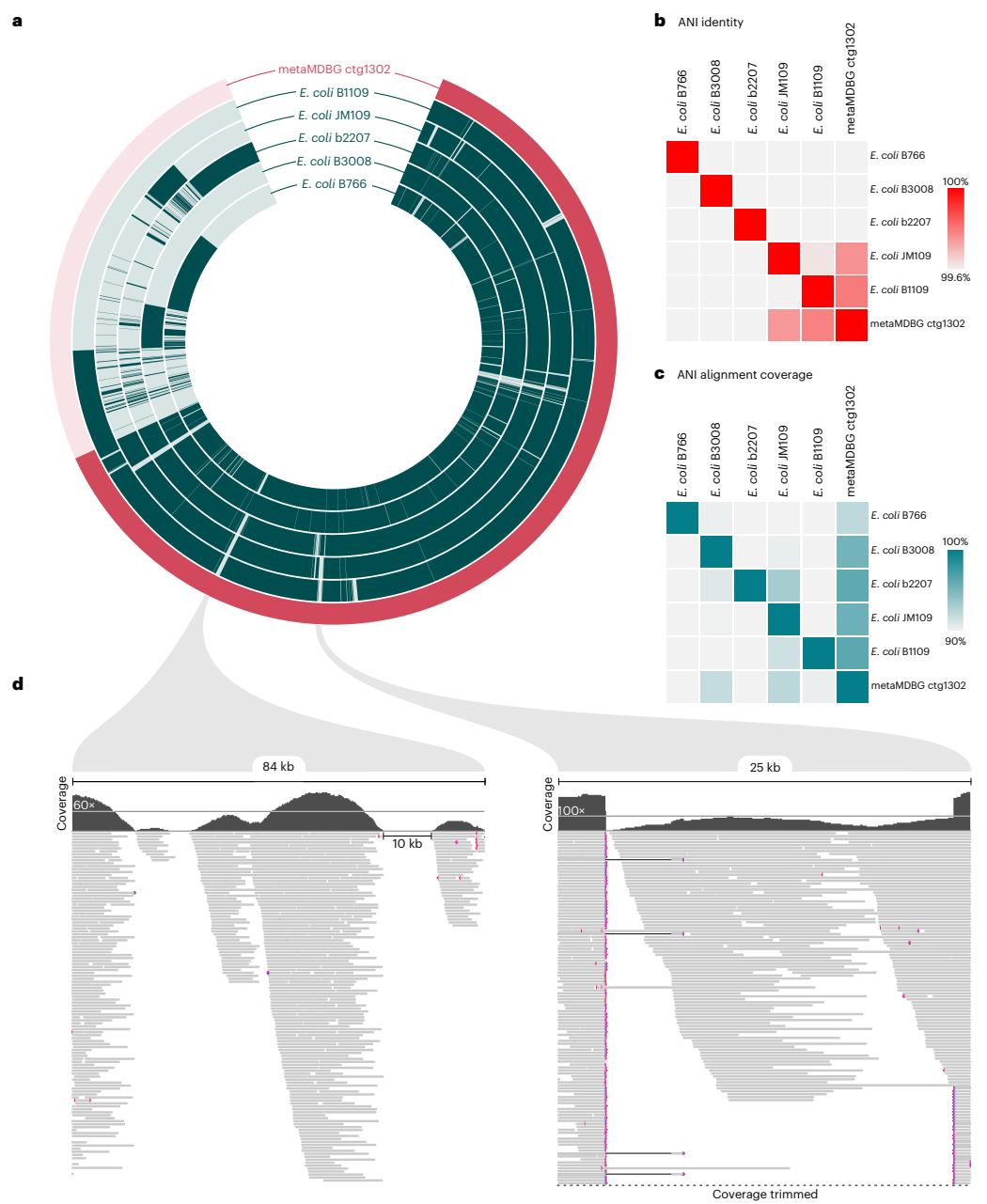
��ϵ�y�u���L�x�L�����M�M�b�|�����о��ˆT�ڡ�Nature Biotechnology���l���о����xȡHiCanu��hifiasm-meta��metaFlye��metaMDBG�Ŀ������M�b���ߣ�������ģ�MȺ�䡢�c������M�ͺ���ӱ���21��PacBio HiFi�������M�л��ʜyԇ��ͨ�^�_�l�_Դ����anvi-script-find-misassemblies�����x�μ����¼������L�x���ڱȌ��^���б�ϵ�y�Էָ�ĬF���״Ό��F�M�b�e�`�ʵľ��_Ӌ����
�P�I���g����������ʹ��minimap2�M���L�x���ccontig�Ȍ���ͨ�^anvi'oƽ�_����contig�����첢�M�л���עጣ�����BLAST��k-mer������C�㸲�w�^�Y��pangenome��������M�������u��Ƕ���w�e�`������ӱ�����������Ⱥϵ���������������˽Y�������m�ԡ�
�M�b�e�`�ձ�����������L�x�L�M�b����
�о��l�F���нM�b���߾����ڸ����Ŷ��x�μ����¼���100%�����Ҹ��w�ȡ�10������metaMDBG�ں���ӱ��Юa���ļ����¼���hifiasm-meta���������������e�`������_ÿ1�|�A����46���e�`���㸲�w�^��>1,000 bp���F��ͬ���ձ飬metaMDBG��5.3%��contig���ڴ���}���h��contig���������@�������g���metaMDBG���ĭh��contig�����77%���ڼ����¼���
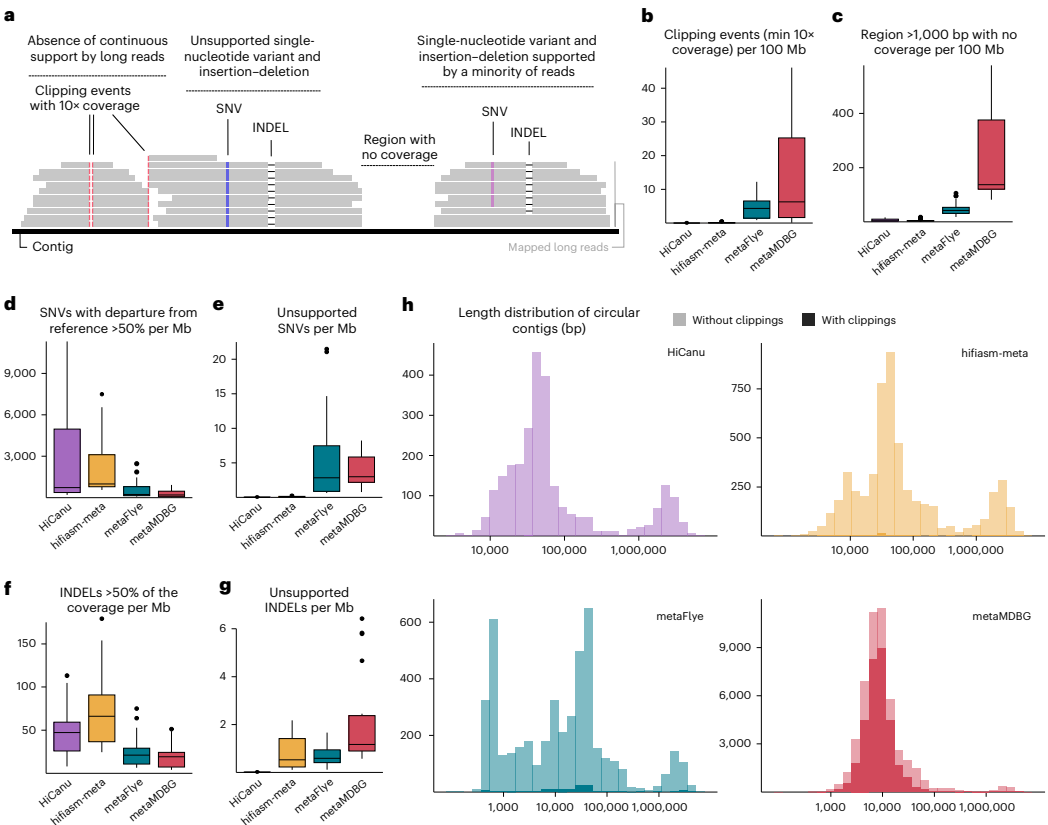
Ƕ���wcontig
�о���ʾ����Ƕ���w�F����metaMDBG���ɵ�contigͬ�r�����V�ž��T���نΰ����T���M�U���T���{���T���С�����δ�|�l���о���7.38 Mb��contigͨ�^�ο�ؐ���Ļ���SCG������ȷ������l�Fƴ�Ӄɂ����Ͼ��ƷNȺ���mȻGC���������w��ͻ׃��ָ�˿��o���R�eǶ���w������Ҏģ����M�о��д���|�����Ƴ�����ҕ��
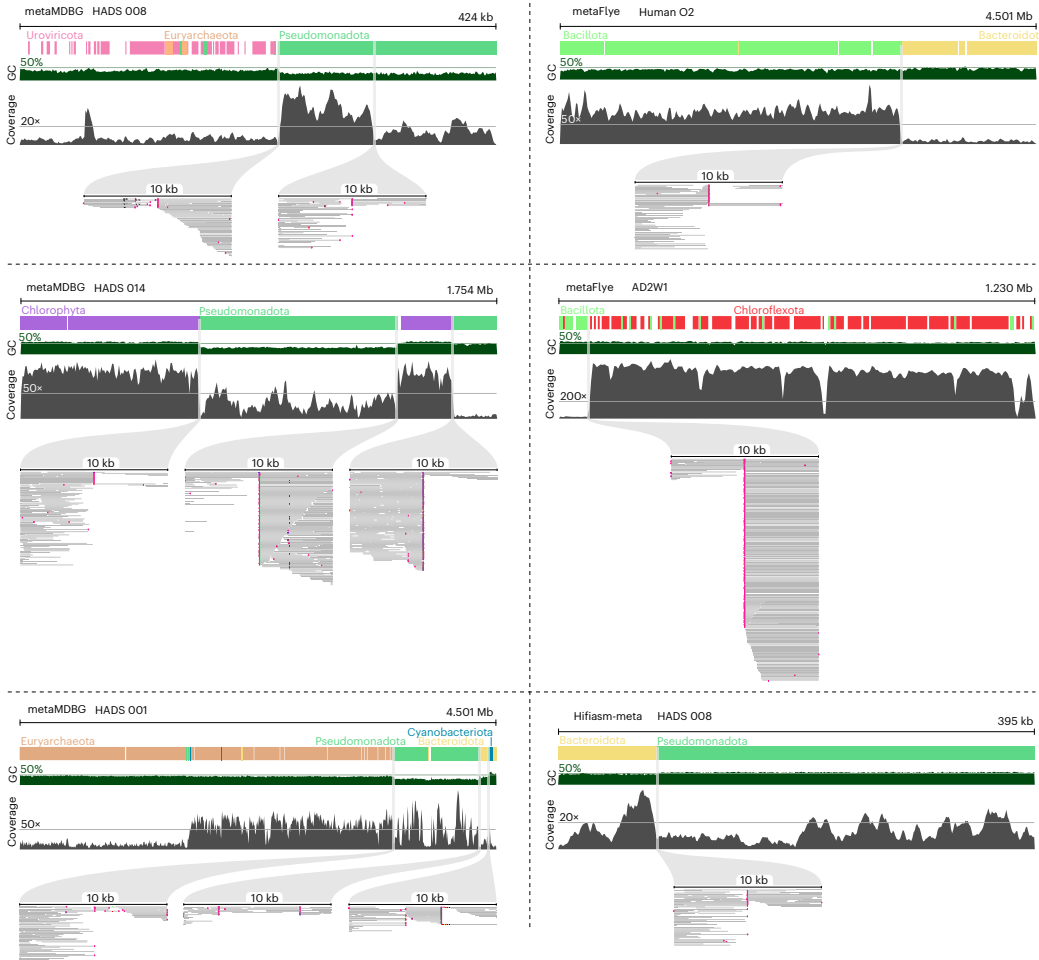
�^��h��
�h��contig���C�ƴ��ڇ��ؿɿ��Ԇ��}��hifiasm-meta�ą��������ؘӱ��Эh���ļ���z���ٻ���Mȱʧ���������P�I���xģ�K�������cλ���D��ø������ͨ�^�O�ñ��غY�x�˜ʣ��h��contig<500 kb�Һ���3�������w���ף����l�FmetaMDBG��̓�٭h������hifiasm-meta��2����metaFlye��4��������e�`���|��/��������M�ؽ��a������Ӱ푡�
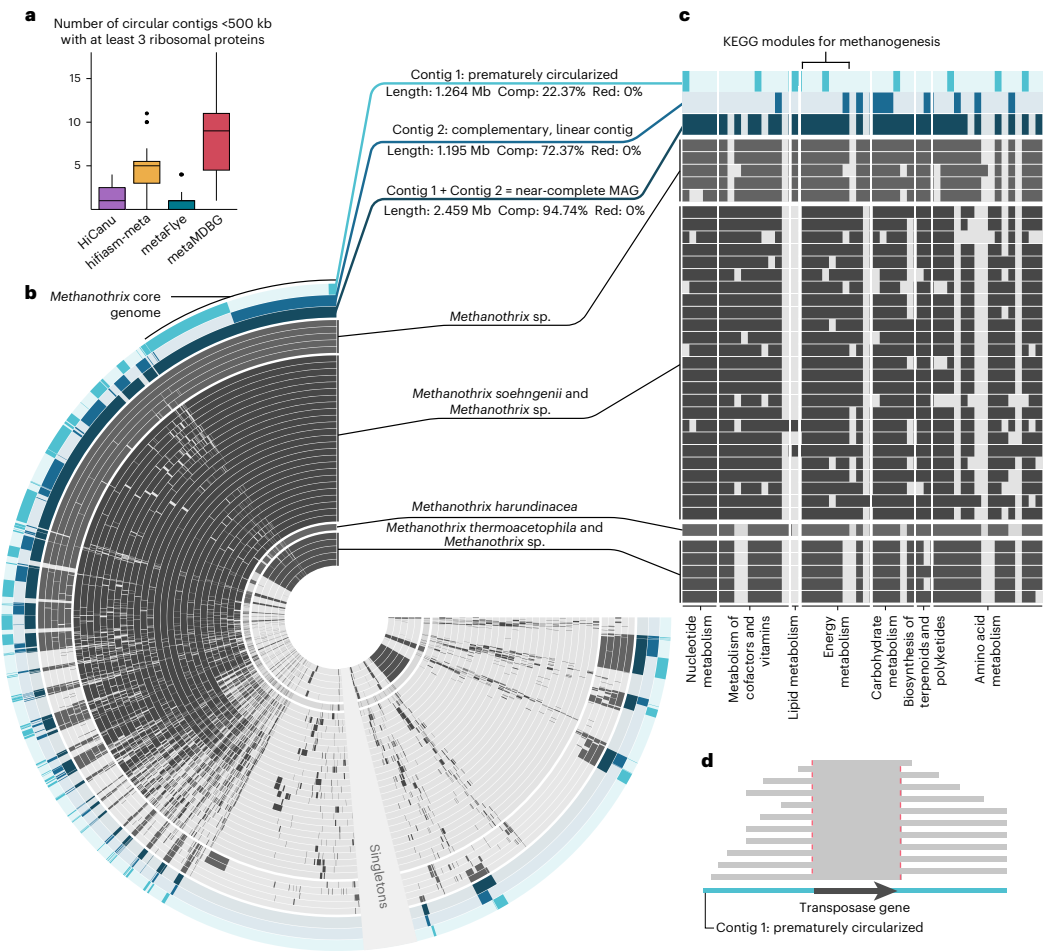
�α����e�`��̓���؏��c��Ӱ����
�о�������N�α��ͽ����e�`��������^���c׃���^�e�`ƴ�ӡ���Ҫ�α������б��ɼ{�������С��o�x��֧�ֵ�̓���؏͡�metaMDBG��metaFlye�����a�����^5,000 bp��"��Ӱ����"����90%��k-mer��k=21����ԭʼ�x����ȱʧ���@Щ�e�`�������_����x��ORF���e�`�A�y��
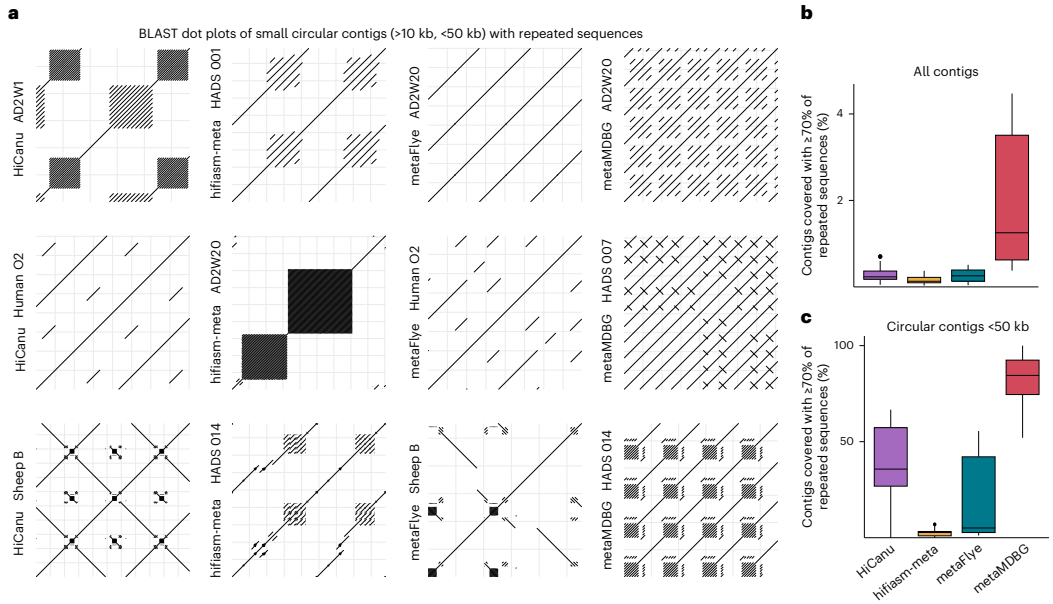
�^���؏�
�ԱȌ������@ʾ��metaMDBG���M�b�ɮa�����^30�f���؏����У�����؏��L���_225,520 bp������ӱ���87%�ĭh��contig��<50 kb����Ҫ���؏����И��ɣ���ʾ�؏�������̓�٭h�����T��֮һ����Ȼ�؏��c�㷨�e�`�ą^����Y�ϸ��w�ȵȶ��ָ�ˡ�
ģ�M�������ľ�����
�о�ָ������ģ�M����������Zymo-HiFi D6331���o����ӳ�挍�ӱ����s�ԡ�hifiasm-meta��ģ�M�����нM�bҎģ������Û��270 Mb vs �A��93 Mb�������ں���ӱ��б��F�������C��ģ�M�����A�y�����ޡ������c�U�������Ϙӱ��ķ����@ʾ��metaMDBG���ɵ�"�h������M"����Ƕ���w��ƽ��������һ���ԣ�ANI�����^�����w����e�`��
���о��������e�`�\�������Ƅ��㷨���M��metaMDBG v1.2��������myloasmͨ�^�����x�μ��з����@�������e�`�ʡ��о��ˆT���{���M�b�㷨���ӏ�����ݔ���x�εĺ��ڼm�e�����ṩ���{���Ć��lʽ�����ԝM�㲻ͬ��������ԓ�������L�x�L�����M�W�r���Ļ���M�ؽ��ɿ��Ԙ����˜ʣ������\�g�������t�W����������M�������|�����Ͼ������h���x��
����ͨ�Ź���̖
֪����I��Ƹ

